In new research this month, doctors say they’ve discovered a new genetic disorder that saps a person’s ability to make antibodies. The likely very rare and treatable condition, first identified in a young boy from Philadelphia, may one day help scientists better understand the immune system.
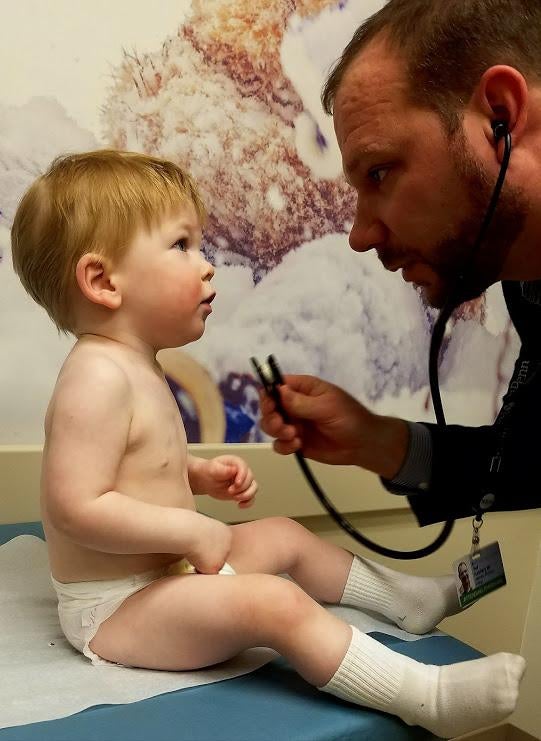
Several years ago, doctors at the Children’s Hospital of Philadelphia (CHOP) came across a mysterious case. Within the first year of his life, patient Luke Terrio had experienced far more infections than normal—infections that were no longer responding to antibiotics and were starting to stunt his development.
The team of specialists quickly surmised that the boy had agammaglobulinemia, a type of immune deficiency where people produce little to no B cells, which help fend off infections. One key function of B cells is producing antibodies, which are especially helpful in neutralizing infections from germs the body has already encountered. But Terrio didn’t appear to have X-linked agammaglobulinemia (XLA), a common form of the condition, as the doctors initially suspected. They soon theorized that his condition was caused by an unknown genetic flaw.
To get to the bottom of this mystery, they sequenced Terrio’s whole exome, the part of our DNA that codes proteins. Eventually, they also compared his exome to others with agammaglobulinemia. And in six of these patients, including Terrio, they found a common link: mutations that hampered the ability to produce a protein important to B cell formation called PU.1. Once they discovered this link, they ran experiments with the gene-editing technology CRISPR, finding that stem cells (taken from donor umbilical cords of unaffected people) edited in the lab to have these mutations began to dysfunction in the same way.
The doctors say the findings of their investigation, published last Wednesday in the Journal of Experimental Medicine, are enough to show that they’ve uncovered a new form of agammaglobulinemia, which they’re calling PU.1 Mutated agammaglobulinemia, or PU.MA. Unlike other forms of the condition, PU.MA. doesn’t seem to be caused by inherited mutations passed down through families, but mutations that arise spontaneously in a developing embryo.
“Based on our experience, we think PU.1 mutations are responsible for ~20% of currently undiagnosed agammaglobulinemia cases,” senior author Neil Romberg, an attending physician with the Division of Allergy and Immunology at CHOP, told Gizmodo in an email. “Agammaglobulinemia is an uncommon disease, so we would expect PU.MA cases to occur in the range of 1 in 1,000,000-7,000,000 live births. So, we think it is quite rare.”
Romberg and his team’s discovery is the first to pin mutations involving PU.1 to human disease. But past animal studies had suggested that mice with these defects would not only develop a weak immune system but also be at higher risk for cancer. That possibility worried his team enough to avoid the standard treatment for agammaglobulinemia for Terrio—regular infusions of replacement antibodies. Instead, Terrio received a living match bone marrow transplant from his older brother Jack.
“I lost some sleep about possible cancer disposition when we first identified Luke’s mutation, and this was one of the primary reasons he got a transplant rather than opting for lifelong antibody replacement therapy,” Romberg said. “Once we identified other PU.MA patients, some in middle age, and realized none had cancer, my concern lessened. We hope this promising trend persists. We will closely watch these patients over time.”
The transplant should allow Terrio to eventually produce a robust supply of antibodies. In the meantime, he’s also receiving regular antibody infusions. Now 4 years old, he’s doing much better and able to run and play like kids his age. And with treatment, patients like Terrio are expected to live long and productive lives, Romberg said.
There are still many mysteries about PU.MA and PU.1 left to be solved. Most of the patients they identified started to become sick within their first year of life, but in at least one patient, the loss of antibodies didn’t seem to start until they reached adulthood—so there may be different ways these mutations are interacting with a person’s environment to cause trouble. Other research has shown that PU.1 is a crucial building block of the immune system and that genetic variations involving it could play a role in other immune-related conditions.
“I suspect PU.MA patients, although very rare, have a lot to teach the world’s scientists about more common disorders like cancer and also inflammatory diseases,” Romberg said. “The PU.MA patients we worked with certainly taught our team a lot about biology and also about perseverance. As a group, they’ve been through a lot.”
More: Some people produce antibodies to opioid drugs, study finds